Vaccine Insights 2023; 2(5), 181–190
PUBLISHED: 13 JUNE 2023
EXPERT INSIGHT
Christopher Ton, Michael A Winters, Raymond Ducoat, Douglas D Richardson, Kristine Fuller, Melissa Hughes
Epidemic and pandemic outbreaks can increase mortality, cause upheaval to healthcare systems, and disrupt global economy and security. Given these threats, it is imperative that there are rapid responses to outbreaks to limit human, social, and economic costs of pandemics. The Ebola virus disease (EVD) epidemic and COVID-19 pandemic posed serious threats to global health, affecting millions to billions of people and disrupting public health services worldwide. Although the viruses associated with EVD and COVID-19 have demonstrated strong infectivity, the high fatality rate of EVD has restricted its spread and prevented it from reaching pandemic level. The responses to the Ebola virus and SARS-CoV-2 outbreaks from manufacturers such as Merck & Co., Inc., Rahway, NJ, USA (MSD) have pushed the boundaries for vaccine development in several areas, including accelerated, parallel clinical and commercial development timelines, implementation of single-use tech-nologies in manufacturing, and engagement with partners and regulatory agencies globally. This review describes how MSD 1) applied the recombinant vesicular stomatitis virus (rVSV) vaccine platform to quickly develop a vaccine for Ebola virus and 2) applied both the rVSV platform and prior knowledge gained from development of the Ebola virus vaccine to rapidly respond to the SARS-CoV-2 pandemic.
Platform process development & manufacturing
Among the various vaccine platforms, live virus vaccines (LVV) are considered the most effective at eliciting life-long cellular and humoral immune responses [1]. However, developing a manufacturing platform for LVV is challenging since different virus families may require different cell substrates, production processes, and purification requirements. LVV can be either attenuated strains or recombinant strains; live recombinant vaccines are replicating viruses that are genetically engineered to carry heterologous antigens. One advantage of live recombinant viruses is that the presentation of heterologous proteins in combination with mimicry of natural infection from live viral vector can generate strong humoral and cellular immune responses without an adjuvant [2]. In the past decade, rVSV has been established as a live recombinant vaccine platform for multiple viral diseases [3].
VSV is a member of the Rhabdoviridae family of negative-stranded RNA viruses and causes non-lethal disease in cattle, horses, and pigs; human VSV infections are rare [4]. rVSV was first developed as a replicating vaccine platform by John Rose and Michael Whitt [5] [6]. Many aspects of the rVSV are advantageous for vaccine development:
- VSV can be propagated to high titers in many cell lines;
- VSV elicits strong cellular and humoral immunity in vivo;
- The VSV-G protein, the major virulence factor of VSV, can be eliminated, thus attenuating the virus and reducing its reactogenicity [7];
- There is a low prevalence of immunity to VSV in most of the general population, making it advantageous to use rVSV as a vaccination platform;
- VSV replicates within the cytoplasm of infected cells and does not integrate into the host genome, reducing the risk of oncogenesis and mutagenesis [8].
Vero cells have been the workhorse for vaccine production over the past 40 years. The cell line was established from cells isolated from a kidney of a normal African green monkey [9]. Vero cells are one of the most common continuous cell lines used for vaccine production; they have been extensively characterized and have gained global acceptance by regulatory authorities [10]. Vero cells do not produce type I interferon in response to viral infections [11], which may explain the susceptibility of these cells to many viruses. This broad susceptibility of Vero cells to many viruses makes them an ideal cell substrate for the development and production of viral vaccines.
Ebola vaccine
On August 8, 2014, the WHO declared the EVD outbreak in West Africa a Public Health Emergency of International Concern [12]. Understanding the urgency of developing an effective vaccine for Ebola virus, NewLink Genetics Corp., in partnership with the US FDA, reached out to MSD to develop an rVSV Ebola vaccine candidate. With extensive internal knowledge of developing LVV, working experience with Vero cells, and scaling up viral vaccine production, MSD partnered with NewLink Genetics Corporation to develop the Ebola vaccine manufacturing process. Leveraging data from the Public Health Agency of Canada, NewLink Genetics and contract manufacturer IDT Biologika, existing literature for rVSV, and extensive internal knowledge of scaling up vaccine production with Vero cells, MSD initiated process development of a robust and scalable manufacturing process.
MSD was challenged with scaling up the existing Ebola rVSV process from 90 to 400 roller bottles to meet Pre-Licensed Patient Access (PLPA) needs. Understanding that the process had to be scaled up quickly, Vero cell expansion experience from the RotaTeq® vaccine was leveraged to develop the Ebola vaccine process. There were two main areas of focus:
- Infection and harvest parameters;
- Scalable downstream unit operations.
To accomplish this, the development team performed repeated cell expansions to generate material to initiate harvest/infection and downstream experiments. Specifically, increased filter surface area, a new tangential flow filter scheme, decreased lumen diameter to maintain shear, and reduced circulation rate was implemented.
Providing significant starting material to these teams was imperative to allow the creation of multiple side-arm experiments to test various process changes simultaneously in parallel experimental arms. This methodology also provided opportunities to complete non-GMP full-scale runs on the upstream process in the pilot plant facility, allowing electronic notebook documentation to later be adapted to production batch records. Experiments were led by a pilot plant operations team, leveraging experience from team members who had previously worked in biologic and vaccine process development areas. The multitude of small-scale purification runs provided hands-on experience to the team that would later be tasked with scale-up for GMP production. Co-locating process development and GMP clinical manufacturing in the same organization with the same scientists eliminated the need for tech transfer.

To accelerate the manufacture of drug substance for Ebola vaccine, the development timeline was drastically compressed (Figure 1). The time from initiation of process development activities at MSD to completion of manufacture for the first batch of GMP PLPA drug substance was 7 months. MSD was able to shorten the development timeline for rapid transfer to manufacturing by executing development and manufacturing scale-up activities in parallel and by implementing single-use technologies. The 400-roller bottle manufacturing process, while not state of the art, was completely disposable end-to-end. Single-use systems provided agility and scalability in a manufacturing facility. Different single-use systems at different scales were installed, commissioned, or removed quickly to meet production requirements. Furthermore, the use of single-use systems reduced manufacturing timelines via the elimination of cleaning validation, clean-in-place, and sterilization-in-place.
Prior to the partnership with MSD, IDT Biologika had initiated a Phase 1 clinical study utilizing material from their existing roller bottle process. In order to use the data from this ongoing study for lot consistency, MSD could not deviate from the roller bottle process to deliver PLPA material. Only changes that supported an increased manufacturing scale were evaluated. For upstream, the process was scaled from 90 to 400 roller bottles to produce the necessary drug substance volume sought for PLPA use. The optimal multiplicity of infection and time of harvest were also determined for the scaled-up process. For downstream, loading studies were performed on the clarification filter to minimize surface area and properly size the filter area needed at larger scale. Range-finding experiments were conducted on the enzyme treatment step in an attempt to reduce the amount of Benzonase® endonuclease used in the process, thereby reducing cost. Temperature studies were conducted to evaluate if simpler, room-temperature manufacturing operations could be utilized. A constant volume ultrafiltration/diafiltration process was implemented to keep process volumes low and reduce manufacturing times. Multiple ultrafiltration filters were evaluated to replace the existing filter, which was not available at the increased scale.
Parallel process development, scale-up, and process transfer to the clinical manufacturing facility also enabled rapid Ebola vaccine development. As each process step was defined, GMP batch records were created by the same engineers, leveraging their experimental knowledge and experience. This brought speed and accuracy to the authoring process. Large-scale roller bottle processes, particularly at the scale demonstrated here, were manual in nature and required intensive hands-on training for execution. The pilot plant operations staff quickly recruited and upskilled new contract staff members to support clinical GMP manufacturing operations. After completing training, these staff members were assigned to help complete experimental work, later transferring these important skills to GMP production.
MSD initiated a Phase 1 clinical trial in fourth quarter 2014, and a Phase 2/3 and consistency lot studies were initiated in February and April 2016. Clinical efficacy data was obtained in June 2016, and ERVEBO® was licensed by the FDA in 2019 [13]. Prior to licensure, the VSV Ebola vaccine was deployed in Guinea in 2015 during the West African Ebola epidemic and the 2018-2020 Democratic Republic of Congo outbreak using the PLPA process, demonstrating 100 and 97.5% efficacy respectively after a single dose [14] [15]. The success of ERVEBO demonstrated the effectiveness of the rVSV vaccine platform in pandemic settings.
COVID-19 vaccine

In response to the COVID-19 outbreak, MSD and the International AIDS Vaccine Initiative (IAVI) applied the rVSV vaccine platform to develop V590, a vaccine candidate for SARS-CoV-2 (Figure 2) [16]. Early integration and real-time data sharing between discovery and process development teams at MSD enabled clone selection for optimal antigenicity and manufacturability. The use of the ERVEBO vaccine production platform also reduced the time required for V590 process development prior to the production of Phase 1 clinical supplies. For example, the ERVEBO upstream roller bottle process, with minor modifications, was leveraged for the production of V590 Phase 1 clinical supplies. The number of roller bottles was increased from 400 to 600 to ensure a sufficient supply of drug substance for Phase 1 clinical trials, and the infection time was reduced by roughly 12−24 hours compared to the ERVEBO process. While several downstream purification process steps were adopted directly from the ERVEBO process, differences between the VSV∆G-ZEBOV-GP and VSV∆G-SARS-CoV-2 viruses required the removal of the ERVEBO protease incubation step with TrypLE™ from the V590 process. This ultimately led to the inclusion of an aseptic, flow-through chromatography step using gamma-irradiated, sterilized Capto™Core 700 resin (Cytiva) to increase clearance of residual host cell proteins. A change to the final drug substance buffer was also incorporated to allow for improved V590 drug product shelf-life.

Though it was recognized that a roller bottle process with aseptic downstream processing would not be used for commercial-scale production due to the large number of anticipated doses for a COVID-19 vaccine, this fit-for-purpose approach allowed for rapid production of Phase 1 clinical supplies. Phase 1 clinical supplies were generated approximately 2 months after V590 clone selection (Figure 3). This is in contrast to traditional preclinical development of vaccines, which usually takes 1−2 years [17].
Other factors besides leveraging the ERVEBO vaccine production platform also enabled V590 process development and production of Phase 1 clinical supplies. The development of multiple Simple Western™ assays allowed for rapid (<1 day) turnaround of analytical results to measure viral and host cell protein levels across downstream processing steps [18]. On-demand potency with rapid plaque and microplaque assays was also quickly established, providing virus infectivity results in less than 48 hours. Project teams were also highly coordinated to align objectives and experimental plans across several workstreams (upstream, downstream, formulation, analytics).
The development time for the COVID-19 vaccine was also shortened by running development and manufacturing activities in parallel. While Phase 1 clinical materials were being manufactured, development and scale-up activities for the commercial-scale production process were executed at the same time, thus reducing cycle times by approximately 18 months. Cross-training was also implemented to ensure efficient process transfer to clinical production, and manufacturing staff were trained on each process step prior to GMP manufacture. This training provided opportunities for the manufacturing team to develop a strong technical knowledge of the production platform. Both Ebola and COVID-19 vaccine production leveraged existing MSD manufacturing facilities with standard unit operations, which enabled rapid process transfer to the clinical manufacture area. In addition, leveraging the existing rVSV vaccine production platform facilitated rapid scale-up and manufacture by utilizing the available validated equipment and GMP-quality raw materials for manufacturing.
To maintain an accelerated timeline, there was significant pre-investment into the development of the commercial-scale production process prior to the availability of Phase 1 clinical results. For commercial-scale production of V590, it was not possible to leverage the Phase 1 roller bottle process for large-scale manufacture. To achieve the number of vaccine doses required to support the pandemic scale, the commercial manufacture process would need approximately 10,000 roller bottles per batch. Thus, we developed a scalable, microcarrier-based bioreactor (2000 L) production process to generate the number of vaccine doses needed for pandemic scale. The 2000 L bioreactor achieved a peak virus titer of ~1.0e+7 plaque forming unit (PFU)/mL [19]. The introduction of the bioreactor process also removed an aseptic control risk inherently associated with roller bottle cultures. To this end, the incorporation of terminal sterile filtration was also considered critical to eliminate aseptic processing and reduce the risk of non-sterile product. The use of a microcarrier bioreactor process and the inclusion of a terminal sterile filtration step for the commercial scale V590 production required substantial process development. Several factors enabled commercial-scale process development to be completed quickly:
- The Vero cell expansion process, which is currently used for MSD’s commercial vaccines and part of its LVV microcarrier process, was leveraged for the V590 commercial scale process so that only the microcarrier N-1 cell expansion step had to be developed to supply a sufficient number of cells for 2000 L bioreactor inoculation;
- Experience from the LVV platform process also enabled a consistent and high-quality supply of Vero cells for infection with VSV∆G-SARS-CoV-2 on a regular schedule. This provided material for downstream process development and production of drug substance for assay and formulation development;
- The utilization of single-use technology, existing equipment, consumables, and raw materials enabled process development experiments to start quickly, increased process flexibility, and allowed for rapid implementation of process changes and demonstration of process iterations [19];
- Processing buffers/media were identified early in development, and the number of buffers used was minimized to reduce the workload required for qualification testing;
- The same type of filters for the clarification (Sartoclean® CA, Satorius) and hollow fiber tangential flow filtration (ReadyToFilter Hollow Fiber Cartridge, 750 kilodaltons nominal molecular weight cutoff membrane, Cytiva) steps that were used for the of manufacture Phase 1 supplies were used in the Phase 3 process. Volumetric loadings were optimized to minimize filter surface area requirements.
While LVV development usually takes approximately 18–24 months from clone selection to implementation of a Phase 3 clinical manufacturing process, the factors above enabled MSD to develop a Phase 3 GMP-compliant 2000 L single-use bioreactor process for V590 in approximately 5 months from clone selection, with Phase 3 clinical supply produced in less than 6 months.
Regulatory interactions
Before ERVEBO approval, emergency use doses were provided to Africa using the IND under Expanded Access protocols. Because this vaccine targeted an unmet medical need, ERVEBO was granted Breakthrough Therapy designation by the FDA and PRIority MEdicines (PRIME) designation by the European Medicines Agency (EMA) [20]. This enabled increased interactions with the regulators (~23 interactions between the EMA and FDA) throughout Biologics License Application and Marketing Authorization Application submission and approval. The applications were submitted using a rolling submission strategy agreed upon with the regulators and also leveraged a collaborative review process with WHO, African VAccine REgulatory Forum (AVAREF), and multiple African countries to ensure approvals were obtained expeditiously where the vaccine was needed most. An overview of expected and actual review periods is shown in Table 1.
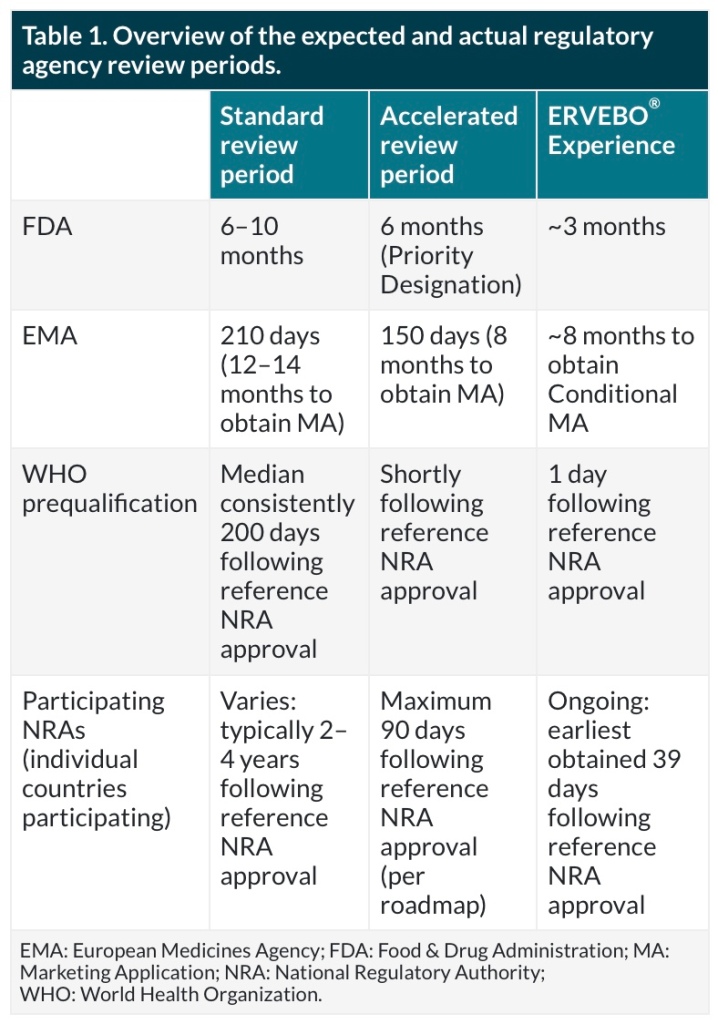
For the COVID-19 vaccine candidate, MSD was able to leverage the ERVEBO roller bottle platform production process to waive preliminary nonclinical studies. Due to the urgency caused by the pandemic, MSD was also able to engage early with agencies to discuss options to accelerate the path to first-in-human. These early engagements included a pre-IND meeting, multiple informal meetings with the FDA and EMA, and Type C written interactions all enabling rapid response and a collaborative sponsor-regulator experience. The early interactions with the FDA enabled the use of a Type V Drug Master File to submit available CMC sections for review earlier than the complete Phase 1 IND package. This allowed the Phase 1 review process to proceed to first-in-human much faster than the normal timeline. After Phase 1, Type C written feedback was also rapidly obtained by submitting written background packages to the IND instead of holding Type C meetings. MSD’s V590 was found to be safe in a Phase 1 clinical trial but was discontinued due to low antibody responses [21].
Expanded access
Prior to ERVEBO approval, the rVSV Ebola investigational vaccine was used to help to contain the outbreaks in the Democratic Republic of the Congo and surrounding countries. Hundreds of thousands of labeled stockpile vaccine doses were deployed through a pre-license access pathway. Pre-license access aims to provide life-saving investigational drugs or vaccines prior to the approval of the drug or vaccine by a regulatory authority. MSD partnered closely with the WHO to align relevant health authority requirements for the use and export/import of rVSV Ebola vaccine into outbreak countries. MSD’s quality control systems were responsible for assessing and approving the WHO’s pre-license access requests and subsequently releasing investigational vaccine lots for use in designated countries. Following release, the MSD logistics organization closely collaborated with specialized pharmaceutical couriers, airlines, and WHO country representatives to seamlessly and routinely deliver vaccine supplies under -70°C dry ice shipment conditions. Extensive pre-license access experience was gained in providing rVSV Ebola vaccine. Lessons learned over time have been employed forward to streamline pre-license access processes and better prepare MSD to respond to future expanded access needs.
Conclusion
MSD’s responses to the Ebola epidemic and SARS-CoV-2 pandemic have demonstrated the benefit of leveraging the rVSV vaccine platform for the rapid development of vaccines. Despite the unprecedented speed of developing these vaccines, opportunities exist for further acceleration of development timelines. As the human population continues to grow and there is habitat destruction, urban development, and increased global travel, the Ebola virus epidemic and SARS-CoV-2 pandemic will not be the last infectious disease outbreaks impacting global human health. Stopping the next emerging pandemic will require utilizing vaccine production platforms and technologies to speed process development and manufacturing scale-up. To ensure a high probability of success for a vaccine candidate in a pandemic, establishing multiple vaccine platforms will be key in developing an effective vaccine quickly. Improvements in regulatory policies and communications to enhance their flexibility without compromising vaccine safety and efficacy are also critical for pandemic preparedness. Long-term strategies for investment into developing new vaccine platforms and application of new technologies for manufacturing infrastructure must be implemented to prepare for accelerated response to future pandemics. Taking these steps for proper preparation will facilitate rapid vaccine development and production to protect society from future public health emergencies.
References
1. Al-Jighefee HT, Najjar H, Ahmed MN, Qush A, Awwad S, Kamareddine L. COVID-19 Vaccine Platforms: Challenges and Safety Contemplations. Vaccines 2021; 9(10), 1196. Crossref
2. Pollard AJ, Bijker EM. A guide to vaccinology: from basic principles to new developments [published correction appears in Nat. Rev. Immunol. 2021 Jan 5]. Nat. Rev. Immunol. 2021; 21(2), 83–100. Crossref
3. Scher G, Schnell MJ. Rhabdoviruses as vectors for vaccines and therapeutics. Curr. Opin. Virol. 2020; 44, 169–182. Crossref
4. Rose JK, Schubert M. Rhabdovirus genomes and their products. In: The Rhabdoviruses (Editor: Wagner RR). 1987; 129–166, Plenum Publishing Corporation. Crossref
5. Lawson ND, Stillman EA, Whitt MA, Rose JK. (1995). Recombinant vesicular stomatitis viruses from DNA. Proc. Nat. Acad. Sci. USA 1995; 92(10), 4477–4481. Crossref
6. Whitt MA. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010; 169(2), 365–374. Crossref
7. Yahalom-Ronen Y, Tamir H, Melamed S et al. A single dose of recombinant VSV-∆G-spike vaccine provides protection against SARS-CoV-2 challenge. Nat. Commun. 2020; 11(1), 6402. Crossref
8. Monath TP, Fast PE, Modjarrad K et al.; Brighton Collaboration Viral Vector Vaccines Safety Working Group (V3SWG). rVSVΔG-ZEBOV-GP (also designated V920) recombinant vesicular stomatitis virus pseudotyped with Ebola Zaire Glycoprotein: Standardized template with key considerations for a risk/benefit assessment. Vaccine 2019; 1, 100009. Crossref
9. Yasumura Y, Kawakita Y. Studies on SV40 in tissue culture-preliminary step for cancer research in vitro. Nihon Rinsho. 1963; 21(21), 1201–1215. Crossref
10. Barrett PN, Mundt W, Kistner O, Howard MK. Vero cell platform in vaccine production: moving towards cell culture-based viral vaccines. Exp. Rev. Vaccines2009; 8(5), 607–618. Crossref
11. Desmyter J, Melnick JL, Rawls WE. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968; 2(10), 955–961. Crossref
12. World Health Organization (WHO), WHO Statement on the Meeting of the International Health Regulations Emergency Committee Regarding the 2014 Ebola Outbreak in West Africa, 2014. Crossref
13. Wolf J, Jannat R, Dubey S et al. Development of Pandemic Vaccines: ERVEBO Case Study. Vaccines (Basel) 2021; 9(3), 190. Crossref
14. Henao-Restrepo AM, Longini IM, Egger M et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 2015; 386(9996), 857–866. Crossref
15. Henao-Restrepo AM, Camacho A, Longini IM et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!) [published correction appears in Lancet. 2017 Feb 4;389(10068):504] Lancet 2017; 389(10068), 505–518. Crossref
16. Saville M, Cramer JP, Downham M et al. Delivering pandemic vaccines in 100 days – what will it take? N. Engl. J. Med. 2022; 387, e3. Crossref
17. Espeseth AS, Yuan M, Citron M et al. Preclinical immunogenicity and efficacy of a candidate COVID-19 vaccine based on a vesicular stomatitis virus-SARS-CoV-2 chimera. EBioMedicine 2022; 82, 104203. Crossref
18. Gillespie PF, Wang Y, Hofmann C et al. Understanding the Spike Protein in COVID-19 Vaccine in Recombinant Vesicular Stomatitis Virus (rVSV) Using Automated Capillary Western Blots. ACS Omega 2023; 8(3), 3319–3328. Crossref
19. Ton C, Stabile V, Carey E et al. Development and scale-up of rVSV-SARS-CoV-2 vaccine process using single use bioreactor. Biotechnol. Rep. (Amsterdam, Netherlands) 2023; 37, e00782. Crossref
20. Wolf J, Bruno S, Eichberg M et al. Applying lessons from the Ebola vaccine experience for SARS-CoV-2 and other epidemic pathogens. NPJ Vaccines 2020; 5(1), 51. Crossref
21. Robbins JA, Tait D, Huang Q et al. Safety and immunogenicity of intramuscular, single-dose V590 (rVSV-SARS-CoV-2 Vaccine) in healthy adults: Results from a phase 1 randomised, double-blind, placebo-controlled, dose-ranging trial. EBioMedicine 2022; 82, 104138. Crossref
Funding declaration:
Development of ERVEBO® was funded in whole or in part with Federal Funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority under Contract Number: HHSO100201500002C. The V590 program has been funded in part with Federal funds from the Department of Health and Human Services, Administration for Strategic Preparedness and Response; Biomedical Advanced Research and Development Authority (BARDA), under Contract No. HHSO100201600031C.
Applying lessons from the Ebola vaccine experience for SARS-CoV-2 and other epidemic pathogens
https://www.nature.com/articles/s41541-020-0204-7.pdf
npj Vaccines volume 5, Article number: 51 (2020)
Abstract
The world is experiencing an unprecedented global pandemic of coronavirus disease 2019 (COVID-19) caused by a novel coronavirus, Severe Acute Respiratory Syndrome-coronavirus-2 (SARS-CoV-2). Development of new vaccines and therapeutics are important to achieve long-term prevention and control of the virus. Experience gained in the development of vaccines for Ebola virus disease provide important lessons in the regulatory, clinical, and manufacturing process that can be applied to SARS-CoV-2 and other epidemic pathogens. This report outlines the main lessons learned by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD) during development of an Ebola Zaire vaccine (ERVEBO®) and looks ahead to critical lessons beyond vaccine development. It highlights focus areas for public-private partnership and regulatory harmonization that can be directly applied to current vaccine development efforts for SARS-CoV-2, while drawing attention to the need for parallel consideration of issues beyond development that are equally important to achieve global preparedness and response goals.
Introduction
First discovered in the Democratic Republic of the Congo in 1976, the highly lethal Ebola virus has infected people in a number of African countries leading to sporadic outbreaks of Ebola virus disease over the past 40 years. While most of the outbreaks have been limited in geographic scope and number of cases, two of the outbreaks over the last 6 years have been large, resulting in major loss of life and socioeconomic disruption in the region. In early 2014, an outbreak of Ebola virus disease caused by Zaire ebolavirus started in West Africa and was declared a Public Health Emergency of International Concern in August of that year. By the time the outbreak ended in 2016, more than 11,000 people had died and more than 28,000 people were infected primarily in three countries, Guinea, Liberia, and Sierra Leone1.
In the decade prior to the West African outbreak, research efforts for biodefense purposes resulted in the identification of some promising Ebola vaccine candidates that protected monkeys from a lethal challenge of wild-type Ebola virus2. These vaccine candidates were not taken into clinical development prior to the West African outbreak for several reasons, which included the inability to demonstrate clinical efficacy in the absence of an ongoing outbreak and lack of interest by the public health and vaccine development community to invest in the lengthy and costly process of vaccine development without a clear demand for an Ebola vaccine2. The West African outbreak changed this perspective and numerous vaccines entered clinical development during the outbreak resulting in the generation of safety and immunogenicity data for many of these novel vaccine candidates and the demonstration of efficacy for one vaccine3.
Development of ERVEBO® (Ebola Zaire Vaccine, Live), a vaccine for the prevention of Zaire ebolavirus disease
ERVEBO® is a live, attenuated, recombinant vesicular stomatitis virus (rVSV)-based, chimeric-vector vaccine, where the VSV envelope G protein was deleted and replaced by inserting only the envelope glycoprotein of Zaire ebolavirus. Extraordinary efforts were made to advance this vaccine candidate through Phase 1, 2, and 3 clinical trials and the data generated in the context of the West African Ebola outbreak has supported its licensure by the US Food and Drug Administration (FDA), conditional authorization by the European Medicines Agency (EMA) and several African countries, along with prequalification by the WHO. The period of 5 years from the start of Phase 1 trials in Oct 2014 to the approval of this vaccine in Nov 2019, was much faster than the typical 10–15 year timeline for vaccine development and approval4. A timeline of the key activities in the development of this Ebola vaccine is summarized in Fig. 1 and described in detail in the following sections of this article. Through this Ebola vaccine development effort a number of learnings have been identified, which are highly relevant for the current vaccine development efforts in response to the COVID-19 pandemic. A summary of the key lessons learned can be found in Fig. 2.
Fig. 1: Timeline of the development of the Ebola vaccine from the start of clinical development through approval.
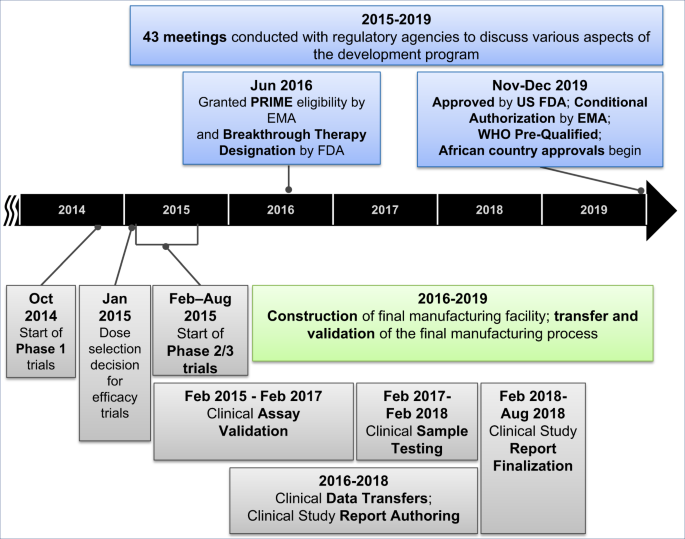
Key clinical and manufacturing activities that were essential for the regulatory approval of the vaccine are noted in this figure.

A strong public-private partnership is needed to bring any vaccine candidate through development during the midst of an infectious disease outbreak
This Ebola vaccine was initially designed as a biodefense vaccine by scientists at the Public Health Agency of Canada (PHAC). Preclinical studies performed by PHAC and their collaborators demonstrated that a single-dose of the vaccine candidate was highly efficacious in animal models. In preparation for potential clinical studies or emergency use, PHAC partnered with IDT Biologika to prepare GMP supplies. Beginning in the autumn of 2014, a diverse set of public-private partners mobilized to collaborate on the evaluation and development of the vaccine candidate. The global collaboration spanned a broad range of organizations, including national governments (such as Canada, the United States, and African countries); various agencies (such as the PHAC, US National Institutes of Health, US Centers for Disease Control and Protection, US Biomedical Advanced Research and Development Authority, US Defense Threat Reduction Agency); field response and non-governmental service organizations (such as Medecins Sans Frontieres); global public health entities (such as the World Health Organization, Wellcome Trust, and Gavi); Universities (e.g., the University of Geneva, Dalhousie University) and private sector companies (such as NewLink Genetics, IDT Biologika, and MSD).
Each of these entities had a specific role in the broad partnership, including conducting preclinical studies, manufacturing Good Manufacturing Practices (GMP) trial materials, conducting clinical trials, and funding portions of the research and development. MSD has a well-established history in manufacturing and clinical development of vaccines, with experience in taking novel vaccines through the regulatory process to approval and providing global access. However, MSD did not have prior experience with Ebola virus nor clinical development expertise in the African countries in which the Ebola outbreak was taking place. Therefore, MSD relied on the expertise of organizations that were already involved in the public health response efforts to implement and complete nearly all the clinical trials. Important lessons from this effort are that leveraging the expertise of different organizations can help to accelerate product development and that broad public-private partnerships require strong leadership and coordination to facilitate cooperation of diverse partners with different expertise and missions.
Furthermore, concerns over evaluation of any investigational product are present in less developed parts of the world and these concerns may be further exacerbated during an outbreak5. Therefore, clear and transparent communication with community leaders and the community at large, ethical review committees, local ministries of health, and other government agencies is critical to ensure that the purpose of the research is well understood and aligned with accepted local and international norms. Failure to do so can result in delays or roadblocks to research on potentially life-saving products.
Role of the marketing authorization holder
Ultimately the responsibility to convert the data generated by all the collaborators into a coherent regulatory submission to support licensure of a product lies with the Marketing Authorization Holder. Recognition and proactive planning for that eventuality can help to accelerate the regulatory submission to support product approval. Examples of these types of activities include the following.
Data integration
MSD’s role in advancing the product to licensure included integrating all the data generated by partners to support the marketing authorization applications. Nine of 13 nonclinical studies, 1 of 2 clinical assay validations, and 11 of 12 clinical trials were performed by partners. MSD needed to obtain all the datasets and reports from these partners to include in the license applications. Data from clinical trials that were performed by partners needed to be migrated into a central database that could be used for data integration, analysis, report-writing, and submission by MSD to regulatory agencies. A lesson learned during this process is to account for the time needed to convert different formats and languages used to collect the data into standard formats required by regulatory agencies, such as use of the Medical Dictionary for Regulatory Activities (MedDRA) and Study Data Tabulation Model (SDTM).
Clinical trial designs
With the clinical trials all being designed and implemented in the context of the outbreak, it was not possible to harmonize the study designs. While this is not an issue a priori for the conduct of the trials, it introduced challenges when assembling the data to support licensure. For example, it was not possible to conduct extensive integration of the safety data for the product because the data collected and methods used were different for each trial. A key lesson learned during this process is that standardization of clinical trial protocols and data collection is preferred, and that to the extent possible late stage clinical trial designs should be defined prior to outbreaks. Another challenge specifically related to conducting trials in response to emerging diseases is that there is a need to build clinical development capacities, laboratory and adverse event reporting infrastructure in outbreak-prone regions. This was a challenge taken on by individual trial sponsors in the affected countries and required significant and sustained support by the trial sponsors and their local partners. Establishing mechanisms to maintain the capabilities established during outbreak response to provide experienced clinical trial sites for future research is another key lesson learned. Examples of this in action include the ongoing partnership between the US and Liberia and the Partnership for Research on Ebola VACcination (PREVAC) consortium established in the wake of the West Africa outbreak.
Clinical assays
A key immunogenicity endpoint assay was developed and validated through a collaborative effort by private and public sector partners. The intent was to provide an assay available to all vaccine developers, which might ultimately allow comparison of different vaccine candidates. While the collaborative effort was successful, balancing the input of numerous partners took time. Consequently, clinical assay validation took two years to complete, resulting in a back-log of samples from clinical trials that needed to be processed. A lesson learned is that whenever possible clinical immunogenicity sample collection should be standardized and assays should be validated prior to the start of late stage clinical trials. This requires knowledge of the target pathogen well in advance of clinical trials, which is a challenge for SARS-CoV-2.
Expanded access use of the vaccine prior to approval
With the rapid collection of efficacy data in support of the vaccine, it was recognized that there was the possibility that new Ebola outbreak(s) could occur ahead of the licensure and availability of licensed doses and that a mechanism was needed to support access to the investigational product. For this scenario, it was necessary to manufacture doses to support outbreak response and establish regulatory frameworks needed to support vaccine deployment (e.g., Compassionate Use, Expanded Access, or Emergency Use). The WHO, MSF, and other groups took the lead in implementing expanded access protocols in African countries, working closely with national governments and ministries of health. Through this effort, more than 300,000 people were vaccinated during the 2018–2020 Ebola outbreak in the Democratic Republic of the Congo6. A lesson learned is that the deployment of an investigational product requires careful consideration and thorough planning.
Manufacturing scaleup
The manufacturing process had to be rapidly scaled-up to handle the needs for emergency use and expanded access programs, in addition to establishing a final manufacturing facility. The approach taken by MSD was to take the initial manufacturing process that was developed at a contract manufacturing organization, scale it up in a clinical manufacturing facility, and transfer it to a commercial manufacturing facility. Technology transfer and the establishment of a new manufacturing facility has many steps and requires significant time to execute (3–4 years is typical for a new vaccine). There is limited understanding outside of vaccine manufacturers and regulators on the rigorous requirements leading to approval of a vaccine manufacturing facility. This broad lack of understanding can lead to misperceptions of “delays” when in fact timelines are driven by the elements required for licensure. For example, although clinical data were available in 2017–2018, the vaccine could not be approved until the commercial manufacturing site was established due to expectations from regulatory agencies that the manufacturing process needs to be validated at the final manufacturing site. A lesson learned is that education is needed on how vaccines are manufactured and the time needed to establish and gain approval for a new manufacturing facility. This is particularly relevant for the COVID-19 outbreak where all aspects of vaccine development are being accelerated at an extraordinary pace with the goal of being able to produce unprecedented quantities of vaccine to address global needs.
Manufacturing site selection for vaccines is a complex decision, which is further complicated when seeking to move fast with incomplete information. The company needed to manage multiple factors, such as existing space, technical capability, infrastructure, and capacity. Site selection also needed to account for the regulations in the country in which the manufacturing site is located and the permits and licenses necessary to support the efforts. The country’s employment environment and labor laws can also impact the ability to hire qualified staff quickly where these factors need to be considered early. A lesson learned is that aggressive timelines are enticing for public health partners, but also require “right-first-time” execution to qualify the facility and manufacturing process, for which the probability of success for a new vaccine is likely medium to low. All parties involved must work to balance the desire to be ambitious (e.g., rapid development timelines) with execution realities and stakeholder expectations. Parallel work and extensive collaboration between manufacturers will be needed in order to successfully bring a SARS-CoV-2 vaccine to the world.
Global and regional harmonization is critical for efficient development and licensure of pandemic vaccines
Regulatory agency collaboration is critical for success
From the start of the West African Ebola outbreak, the US FDA, EMA, and Health Canada worked closely with each other and with the National Regulatory Authorities of the impacted West African countries, sharing information about candidate vaccines that were being tested and reviewing the clinical protocols, available data, and benefit-risk profiles. Frequent conversations with manufacturers helped the agencies expedite the start of clinical trials simultaneously in different countries. A lesson learned is that existing preclinical data and availability of clinical supplies afforded the opportunity to start Phase 1 trials rapidly during the West African Ebola outbreak.
Clinical data requirements for licensure need to be clearly defined
Placebo-controlled, randomized, double-blind studies are typically used to demonstrate the efficacy and safety for new vaccines. However, in the midst of the Ebola outbreak, some countries considered it unethical to administer placebo to at-risk individuals. Different study designs were implemented for the Phase 2/3 trials and a cluster-randomized trial conducted by the World Health Organization in Guinea was the only trial that demonstrated efficacy7. Efficacy data from this trial, together with safety data from 12 trials and immunogenicity data with validated assays, were accepted for registration by regulatory agencies. A lesson learned is that the regulatory pathway to licensure for pandemic vaccines need to be clearly defined. Novel trial designs might need to be implemented in order to maximize the possibility of evaluating efficacy during a waning outbreak, and alternative endpoints, such as using immune responses as a surrogate for efficacy, could be used to gain accelerated approval or conditional marketing authorization.
Frequent regulatory agency interactions are important
Based primarily on interim clinical efficacy data, the manufacturer applied for and was granted access to expedited regulatory pathways, such as Priority Medicines (PRIME) status from EMA and Breakthrough Therapy Designation (BTD) from FDA, which helped to obtain alignment on data requirements for product approval using frequent meetings. Under BTD, the FDA accepted rolling submissions of portions of the Biologics License Application (BLA), which resulted in approval in December 2019, three months before FDA’s target approval date. The FDA determined that a program specific advisory committee meeting was not needed. Similar frequent discussions with EMA under PRIME status, enabled the company to gain alignment on key aspects of the development program quickly.
WHO leadership is needed to obtain rapid global approvals
WHO is a recognized global public health leader with regional offices in many countries, and established connections with the African Vaccines Regulatory Forum (AVAREF). In order to accelerate vaccine access to African countries, the World Health Organization’s Prequalification Team (WHO-PQT) in collaboration with the EMA and AVAREF developed an innovative facilitated process (roadmap) for decision making on the acceptability of the vaccine for registration8. This allowed the company to make simultaneous submissions to EMA, WHO-PQT, and regulatory authorities in 14 African countries, with EMA acting as the reference agency. Following submission, the WHO-PQT and members from AVAREF, which represented the 14 African countries, were invited to participate in teleconferences between MSD and EMA and in the inspection of the manufacturing facility. After a positive opinion from EMA in October 2019, the European Commission granted a conditional Marketing Authorization on November 11th. Within 36 h, the WHO granted prequalification. One month later, African countries started to approve ERVEBO®, starting with Burundi and the Democratic Republic of the Congo. A lesson learned is that strong collaboration and willingness to share information with everyone involved resulted in registration nearly-simultaneously in the countries that needed the Ebola vaccine. For SARS-CoV-2 vaccines, regulatory agencies should prepare for the simultaneous approval of candidates in multiple countries. WHO’s leadership will be needed to facilitate innovative review processes and collaborative mechanisms to expedite approvals.
Recombinant viral vaccines need to overcome additional regulatory hurdles
Since this Ebola vaccine was a recombinant virus, some countries required a detailed environmental risk assessment prior to the start of clinical trials and as part of the marketing authorization application. In some countries, the vaccine was considered a Bio-Safety Level 2 organism, requiring special handling and permits for the manufacturing and testing sites. These permits took a significant amount of time to obtain, which prevented rapid transfers of material. Samples and technology, which even included parts of the marketing authorization application, required licenses for export because this vaccine is made from the genetic sequence of two viruses that are considered “dual-use” agents and subject to trade controls. A lesson learned is that these regulations led to delays and additional costs incurred in starting clinical trials, manufacturing, testing, and shipments. For manufacturers of SARS-CoV-2 vaccines, countries should consider waiving these requirements, or waiving the fees and expediting these processes.
Harmonized labeling and packaging requirements would facilitate pandemic vaccines distribution
After approval, companies face challenges with product distribution due to the need to follow country-specific product labeling regulations covering package inserts, cartons, and artwork in addition to serialization requirements. This causes a supply chain issue for a vaccine product intended to be placed in a stockpile and diverted quickly to any country. A lesson learned is that these heterogenous requirements counter the goal of flexibility, speed, and cost-efficiency relevant to emergency preparedness, and there is an urgent need for harmonized solutions. Electronic labeling and Quick Response (QR) codes could be a possible solution, but in most countries, legislation does not currently exist to support this option. There is an urgent need to find a solution to this problem for pandemic vaccines.
Beyond vaccine development
In addition to considering lessons related to vaccine development for epidemic preparedness, it is equally important to consider the unique challenges and opportunities related to sustainable manufacturing, supply, access, and delivery of these vaccines at the scale and speed needed to achieve preparedness and response goals. Such challenges include right-sizing manufacturing capacity and production while having to accommodate lead-times, managing uncertain demand-and-supply dynamics, ensuring equitable allocation and access, and achieving operational and economic sustainability for all partners. Producing and supplying any vaccine is inherently complex. It is exponentially more complex when there are high levels of uncertainty and unpredictability across nearly every dimension of the program: unpredictable disease, unpredictable and relatively low demand, unpredictable stakeholders and customers, unpredictable timing of need, and unpredictable geographies in which needs might arise. Policy, program, and system innovation—in parallel to research and development innovation—needs to be addressed to realize the full promise of innovative, new vaccines in advancing global public health preparedness.
Conclusions
Emergency preparedness and response vaccines, such as ERVEBO®, are critical tools in the arsenal against pathogens that can cause pandemics. Public-private partnerships are a powerful and effective approach to develop these vaccines. Setting clear roles, expectations, and accountability enables each partner to bring their respective strengths to the effort. Shared purpose, trust, flexibility, and cooperation are critical to ensure success.
As highlighted in this report and in another recent article9, the variability and complexity in regulatory processes and requirements interfere with the speed, flexibility, and efficiency needed to prepare for and respond to public health emergencies. There is an urgent need for global regulatory harmonization and standardization in the approach to the development of vaccines for emergency preparedness. Regulatory agencies need to work together to remove roadblocks and put solutions in place to allow the rapid global development of vaccines for SARS-CoV-2 and other emerging pathogens.
References
- World Health Organization. Ebola Situation Report—March 30, 2016. https://apps.who.int/ebola/current-situation/ebola-situation-report-30-march-2016 (2020).
- Feldmann, H., Feldmann, F. & Marzi, A. Ebola: lessons on vaccine development. Annu. Rev. Microbiol. 72, 423–446 (2018).Article CAS Google Scholar
- Higgs, E. et al. Accelerating vaccine development during the 2013-2016 West African Ebola virus disease outbreak. Curr. Top. Microbiol. Immunol. 411, 229–261 (2017).CAS PubMed Google Scholar
- International Federation of Pharmaceutical Manufacturers & Associations. The complex journey of a vaccine. https://www.ifpma.org/wp-content/uploads/2019/07/IFPMA-ComplexJourney-2019_FINAL.pdf (2020).
- Folayan, M. O., Peterson, K. & Kombe, F. Ethics, emergencies and Ebola clinical trials: the role of governments and communities in offshored research. Pan. Afr. Med. J. 22, 10 (2015).PubMed PubMed Central Google Scholar
- World Health Organization. Ebola virus disease Democratic Republic of Congo: External Situation Report 94. https://www.who.int/emergencies/diseases/ebola/drc-2019/situation-reports (2020).
- Henao-Restrepo, A. M. et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 389, 505–518 (2017).Article CAS Google Scholar
- World Health Organization. Roadmap for introduction and roll out of a licensed Ebola vaccine. https://www.who.int/medicines/news/2019/roadmap_for_intro_roll_out_licensed_ebola_vaccine/en/ (2020).
- Lurie, N., Saville, M., Hatchett, R. & Halton, J. Developing Covid-19 vaccines at pandemic speed. N. Eng. J. Med. 382, 1969–1973 (2020).Article CAS Google Scholar
Acknowledgements
We thank all team members and management for their tireless efforts and contributions to this program. For their partnership and dedication, we thank external partners, collaborators, and funding organizations in addition to the study volunteers and study investigators. The authors also thank the WHO, FDA, EMA, AVAREF, and regulatory agencies that participated in the collaborative review procedure. We would also like to thank Karyn Davis of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA for editorial assistance.
Author information
Authors and Affiliations
- Merck & Co., Inc., Kenilworth, NJ, USAJayanthi Wolf, Samantha Bruno, Michael Eichberg, Risat Jannat, Sharon Rudo, Susan VanRheenen & Beth-Ann Coller
Contributions
J.W. wrote the first draft of the manuscript. All co-authors provided critical inputs and revised the manuscript. All authors agreed to final submission.

Hinterlasse einen Kommentar